Son iguales los genéricos que los fármacos de marca?

Pedro Corsino Fernández Vila, MD
(Artigo em Castelhano)
Fármacos genéricos
Un medicamento genérico es todo medicamento que tenga la misma composición cualitativa y cuantitativa en principios activos y la misma forma farmacéutica que el medicamento de referencia y cuya bioequivalencia haya sido demostrada por estudios adecuados de biodisponibilidad.
A su vez la biodisponibilidad se define como la cantidad y la velocidad a la que el principio activo se absorbe a partir de una forma farmacéutica y llega al lugar de acción (biofase). Habida cuenta que el principio activo está en equilibrio entre la circulación sistémica y el lugar de acción, se asume que los parámetros medidos en sangre del medicamento son representativos de su biodisponibilidad. La biodisponibilidad se evalúa mediante parámetros farmacocinéticos tales como: el área bajo la curva (ABC o AUC, por sus siglas en inglés), la concentración máxima alcanzada (Cmáx) y el tiempo en alcanzar la concentración máxima (Tmáx).
La bioequivalencia es la comparación entre la biodisponibilidad de un fármaco en estudio y la biodisponibilidad de otro fármaco tomado como referencia. Se acepta que el producto en estudio es bioequivalente con el de referencia cuando sus valores (especialmente el AUC) se encuentran dentro del intervalo de confianza del 90 % (80-125 %).
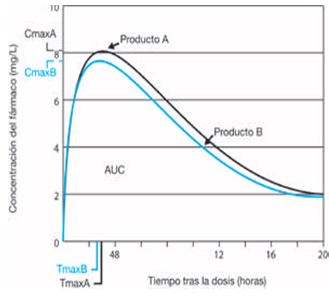
Para llevar a cabo los estudios de bioequivalencia, una vez administrado el fármaco, se realizan extracciones de sangre a distintos tiempos (según la semivida de eliminación del fármaco) y se determinan las concentraciones del principio activo en sangre lo que va a permitir calcular los parámetros cinéticos de biodisponibilidad.
Medicamentos de acción local o tópica
Un medicamento de acción local o tópica es un producto que se aplica localmente y se asume que ejerce su efecto en el lugar de aplicación. Los efectos sistémicos, si existen, deben ser considerados como efectos secundarios. Ejemplos de fármacos de acción local son los productos dermatológicos (cremas, pomadas, etc.), los productos inhalados, las gotas oftálmicas y óticas, los productos nasales y los productos aplicados en las cavidades oral, vaginal y rectal con efecto local.
El principal problema para el desarrollo de genéricos de medicamentos de acción local o tópica es que no se pueden realizar estudios de biodisponibilidad, basados en la comparación del perfil de concentraciones plasmáticas alcanzadas por el fármaco de referencia y la formulación genérica porque… el efecto se produce a nivel de una superficie mucosa o de la piel y no se alcanzan concentraciones plasmáticas lo suficientemente altas que permitan la determinación de la concentración del fármaco, y si se alcanzan, éstas son reducidas e irregulares.
La bioequivalencia, en el caso de las gotas oftálmicas, los esprays nasales y las soluciones cutáneas, es aceptada si el fármaco tiene el mismo tipo de solución (acuosa u oleosa) y contiene la misma concentración de la sustancia activa que el producto de referencia. Se aceptan diferencias menores en la composición de los excipientes siempre que las propiedades farmacéuticas del fármaco en estudio y del fármaco de referencia sean idénticas o esencialmente similares. Cualquier diferencia cualitativa o cuantitativa de los excipientes debe justificarse de forma satisfactoria en relación con la influencia sobre la equivalencia terapéutica. El método y forma de administración debe ser el mismo que el del producto ya aprobado.
La Food and Drugs Administration de EEUU (FDA)1 exige que el fármaco en estudio tenga los mismos ingredientes activos e inactivos y la misma concentración que el producto de referencia. No se aceptan diferencias en la composición de los excipientes. Sin embargo, un laboratorio puede solicitar la aprobación de un fármaco que difiere del original, para lo cual debe identificar y caracterizar las diferencias y proporcionar información demostrando que las diferencias no afectan a la seguridad del fármaco2. El criterio de la FDA para determinar la equivalencia terapéutica es, básicamente, tener equivalencia de formulación.
¿Por qué los fabricantes de genéricos no copian exactamente el fármaco original? La respuesta en una palabra: patentes. Cada fármaco lleva asociadas, como media, cinco o seis patentes. Los fabricantes de genéricos, para evitar infringir las patentes, cambian de forma intencionada el conservante o el pH aunque en realidad nunca llegan a saber con exactitud cómo se fabrica el producto original. Algunos laboratorios producen el fármaco original y el genérico. Merck&Co., Inc. manufacturan el Cosopt original y el genérico por lo que cabe suponer que, al menos en estos casos, los fármacos son exactamente iguales.
De todo ello deriva una pregunta clave: ¿por qué la FDA exige, frente a la Agencia Europea del Medicamento, que todos los componentes activos e inactivos de un fármaco genérico oftálmico sean absolutamente iguales a los del fármaco de referencia?
La FDA1 considera que: 1) Cambiar los excipientes o componentes inactivos de un fármaco puede tener un efecto significativo tanto en la eficacia como en la seguridad del fármaco. 2) Los cambios en el conservante o en la concentración del conservante pueden influir en la penetración corneal del principio activo y 3) Los cambios en los demulcentes o agentes de la tonicidad pueden modificar el tiempo de contacto de la solución con la superficie corneal y por tanto alterar la absorción del principio activo en solución.
Las suspensiones, geles, emulsiones y pomadas oftálmicas y al contrario de lo que ocurre con las soluciones, pueden verse significativamente alteradas aunque los componentes activos e inactivos del fármaco sean cuantitativa y cualitativamente los mismos, por variaciones en los procesos de fabricación (pulverización, distribución del tamaño de las partículas u orden de la mezcla).
Los criterios que sigue la FDA para determinar la eficacia clínica en estos fármacos varían con cada grupo farmacológico y se basan en los parámetros o medidas que fueron utilizados para establecer la eficacia del fármaco innovador. Por ejemplo, en el caso de los fármacos hipotensores oculares, se determina la equivalencia si la diferencia entre los dos fármacos es inferior a 1,5 mm Hg (intervalo de confianza del 95%) en todas las determinaciones horarias. En este mismo caso, en el ensayo clínico se han de realizar determinaciones de la presión intraocular (PIO) basal y a la 1.ª, 6.ª y 12.ª semana, al menos en las horas consideradas pico y valle para ese principio activo.
En la actualidad hay fármacos genéricos oftálmicos de prácticamente todos los grupos terapéuticos. Así, hay: antiinfecciosos, corticoides, antiinflamatorios no esteroideos (AINE), antiglaucomatosos, combinaciones fijas, antialérgicos, midriáticos /ciclopléjicos y anestésicos.
La importancia del excipiente
Tabla 1. Porcentaje de principio activo y excipientes en diferentes colirios antiglaucomatosos
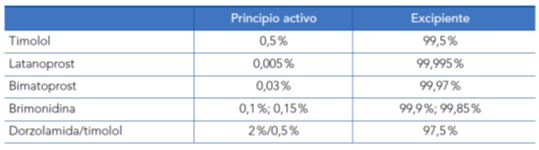
Si más del 95 % de un colirio lo constituyen los excipientes, su importancia es evidente3. Un colirio contiene múltiples excipientes: 1) vehículo líquido acuoso o más raramente oleoso. 2) conservantes. 3) ajustadores del pH. 4) antioxidantes. 5) viscosizantes. 6) buffers. 7) ajustadores de la tonicidad4.
El objetivo primario de los conservantes es inhibir la contaminación bacteriana. Algunos conservantes tensioactivos también contribuyen a que fármacos más lipofílicos, como los análogos de las prostaglandinas, permanezcan en solución.
La importancia del conservante puede verse en que la sola sustitución de un conservante, BAK, por otro, Purite®, permite que un fármaco alcance concentraciones más altas en humor acuoso con la misma concentración (brimonidina al 0,15 %) y similares a las del mismo fármaco con una concentración superior (brimonidina-BAK al 0,20 %)5. La razón es que con un conservante como el BAK es difícil subir el pH de una solución más allá de 6,7/6,8, mientras que con el conservante Purite®, el pH puede alcanzar casi un 7,8. Así cambiando el conservante y subiendo el pH fue posible mejorar la penetración de la brimonidina a través de la córnea e incrementar su concentración en el humor acuoso.
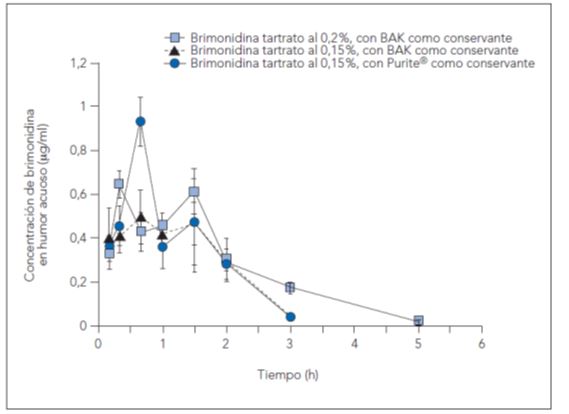
Figura 2. Concentraciones de brimonidina en humor acuoso en el tiempo.
Fuente: Dong JQ, Babusis DM, Welty DF, Acheampong AA, Tang-Liu D, Whitcup SM. Effects of the preservative Purite® on the bioavailability of Brimonidine in the aqueous humor of rabbits. J Ocul Pharmacol Ther. 2004;20(4):285-92.
Por su parte los agentes que modifican la viscosidad aumentan el tiempo de contacto del fármaco con la superficie ocular y mejoran la absorción. Al aumentar el tiempo de contacto, reducen la absorción nasolagrimal y mejoran la seguridad sistémica. A su vez pueden modificar la capacidad de un principio activo de permanecer en solución y estabilizan la interacción con la película lagrimal, lo cual influye en la tolerancia.
En este sentido, Meseguer et al. (1996)6 realizaron un estudio en voluntarios humanos donde se comparó el tiempo de contacto con la superficie corneal de varias formulaciones de pilocarpina con distintos viscosizantes: sistema de transición de fase (gellan gum, Gelrite®), un heteropolisacárido (xanthan gum) y diferentes polímeros habituales usados como solución control: hidroxietilcelulosa, hidroximetilcelulosa o polivinilalcohol. Se instiló una gota de pilocarpina (25 μl) de cada una de las formulaciones en un solo ojo a intervalos de una semana. El tiempo de contacto se determinó por gammaescintigrafía usando como marcador tecnecio 99 (Tc-99m DTPA). Un minuto después de la instilación, sólo permanecía en contacto con la superficie ocular el 23 % de las formulaciones de control frente al 77 % del xanthan gum o el 82 % del Gelrite®. A los 21 minutos de la instilación, permanecían en contacto un 12 % de la solución de control (polímero), el 25 % de la solución de xanthan gum y el 39 % de la solución de Gelrite®.
Tabla 2. Comparación estadistica de la actividad residual sobre la superficie ocular de diferentes soluciones oftálmicas que contenían nitrato de pilocarpina al 0,5%. Primer grupo de voluntarios (media+- DE).
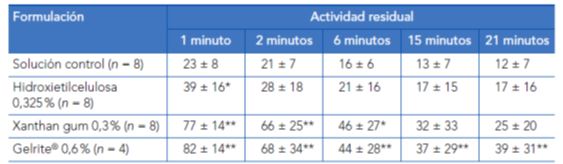
*p<0,05;**p<0,01 (la significación de las diferencias respecto a la solución control se evaluó mediante la prueba de la t de Student).
Fuente: Meseguer G, Buri P, Plazonnet B, Rozier A, Gurny R, Gamma scintigraphic comparison of eyedrops containing pilocarpine in healthy volunteers. J Ocul Pharmacol Ther. 1996 Winter;12(4):481-8.
Dickstein et al. (1996)7 realizaron un estudio cruzado, de distribución aleatoria, doble ciego, en el que compararon los efectos del timolol al 0,5 % en solución acuosa (una gota cada 12 h) y en vehículo de gellan gum (Gelrite®) (una gota cada 24 h) sobre el ejercicio en 42 varones de mediana edad (58 años, intervalo: 55 a 65 años). Los sujetos se ejercitaron al máximo en un cicloergómetro, cuatro veces, a intervalos de 10 días.
Las concentraciones de timolol en suero fueron de 0,91 ± 0,51 ng/ml para la solución de timolol y de 0,71 ± 0,46 ng/ml para el timolol en vehículo de gellan gum. El cambio de la frecuencia cardíaca en reposo fue de -1,8 ± 9,3 lat/min para el placebo, -11,0 ± 9,6 lat/min para la solución de timolol, y de -8,5 ± 7,5 lat/min para el timolol en vehículo de gellan gum. El cambio de la frecuencia cardíaca en ejercicio fue de -0,1 ± 7,3 lat/min para el placebo, -15,6 ± 5,6 lat/min para la solución de timolol, y de -11,9 ± 8,0 lat/min para el timolol en vehículo de gellan gum. Estas diferencias son estadísticamente significativas.
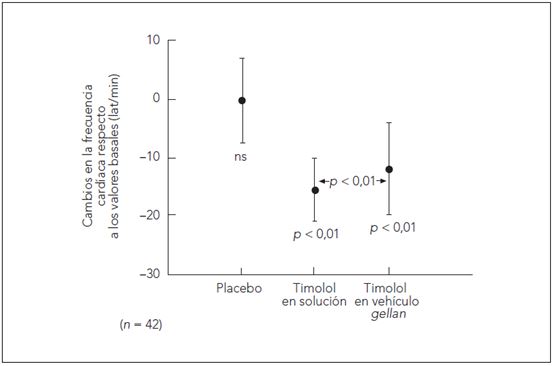
Figura 3. Diferencias en la frecuencia cardíaca (lat/min +-DE) durante el ejercicio. Cambios en la frecuencia cardiaca tras la instilación de placebo, timolol en solución y timolol en vehículo de gellan comparado con la frecuencia basal.
Fuente: Dickstein K, Hapnes R, Aarsland T. Comparison of aqueous and gellan ophthalmic timolol with placebo on the 24-hour heart rate response in patients on treatment for glaucoma. Am J Ophthalmol. 2001;132(5):626-32.
Aunque ambos fármacos redujeron la frecuencia cardíaca de forma significativa tanto durante el período de reposo como en ejercicio, la solución con vehículo de gellan gum lo hizo de forma significativamente menor. La reducción significativamente menor de la frecuencia cardíaca se relaciona con la menor absorción sistémica y, en consecuencia, menor concentración sérica de timolol (significativamente estadística) con el vehículo de gellan gum.
La temperatura influye sobre la concentración de los principios activos contenidos en un colirio. Kahook et al. (2012)8 realizaron un estudio sobre los efectos del calor en distintas formulaciones después de 30 días, en un intento de reproducir la duración de un envase con un uso normal. Se estudiaron las concentraciones de diferentes principios activos (latanoprost, timolol y dorzolamida), así como de cloruro de BAK en distintos colirios de marca y genéricos, tras la exposición a temperaturas de 25 y 50 ºC durante 30 días.
Todos los fármacos, de marca y genéricos, mostraron una reducción de la concentración de principios activos y BAK cuando se expusieron a temperaturas superiores a las marcadas en el prospecto. Las dos formulaciones genéricas de latanoprost contenían una concentración de principios activos que eran un 10% superiores a lo marcado. Las dos formulaciones de latanoprost genérico tuvieron una disminución significativa de la concentración del principio activo tras la exposición a temperaturas de 25 y 50ºC durante 30 días. Las formulaciones genéricas de latanoprost perdieron más del 10 % de la concentración media de su principio activo a una temperatura en el límite superior de estabilidad indicado en el prospecto y durante el tiempo esperable de uso clínico. El aumento de la concentración del principio activo observado en el momento basal puede ser una estrategia para compensar la tendencia conocida de esos fármacos a degradarse en el tiempo a niveles por debajo de su dosis óptima, incluso a temperatura ambiental. La combinación de timolol-dorzolamida, tanto en el fármaco de marca como en las formulaciones genéricas, es relativamente resistente a la degradación.
Los envases de las formulaciones genéricas de ambos fármacos tenían una mayor cantidad de partículas en comparación con los fármacos de marca. El número medio de partículas aumentó significativamente tanto en los fármacos de marca como en los genéricos después de la exposición a 50º C durante 30 días. No se conoce la naturaleza exacta de las partículas. Así, mientras algunas tienen un aspecto sólido, otras parecen de origen fibrilar. El origen de las partículas es desconocido y puede tratarse de contaminantes, precipitados de los principios activos o material del contenedor. Se desconoce si las partículas pueden tener repercusión clínica en la superficie ocular tras su uso habitual.
Tabla 3. Número de partículas (por milimetro de volumen) de diámetro superior a 1 µm en el momento basal y después de ser sometidos a estrés térmico
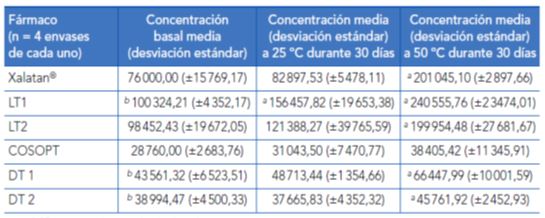
* p < 0,05 comparado con el valor basal.
** p < 0,05 comparado con el valor basal de la marca registrada.
DT, dorzolamida-timolol; LT, latanoprost.
Fuente: Kahook MY, Fechtner RD, Katz LJ, Noecker RJ, Ammar DA. A comparison of active ingredients and preservatives between brand name and generic topical glaucoma medications using liquid chromatography-tandem mass spectrometry. Curr Eye Res. 2012;37(2):101-8.
Los medicamentos genéricos e innovadores pueden diferir asimismo en la forma del envase, en la viscosidad, en la tensión superficial y en el volumen instilado en cada gota. Mammo ZN et al. (2012)9 realizaron un estudio cuyo objetivo era determinar si había diferencias en la forma del envase, viscosidad, tensión superficial y volumen instilado en cada gota entre medicamentos genéricos e innovadores comercializados en Estados Unidos y Canadá. Los colirios de Timoptic XE© de Estados Unidos y Canadá son diferentes, de forma estadísticamente significativa, de los colirios genéricos Timolol GFS© (Estados Unidos) y Timolol Maleate EX© (Canadá), en el volumen instilado en cada gota, viscosidad, tensión superficial y diámetro del orificio del envase.
Los autores concluyen que los colirios genéricos Timolol Maleate EX© (Canadá) y Timolol GFS© (Estados Unidos) liberan aproximadamente tres quintas partes y dos tercios, respectivamente, de la dosis terapéutica diaria cuando se comparan con el medicamento de referencia, específico de cada país, Timoptic XE© y consideran que para que un fármaco genérico pueda ser considerado intercambiable con el fármaco de referencia deberían tenerse en cuenta y someter a normas de regulación más estrictas los denominados componentes inactivos y conservantes de un determinado medicamento.
Tabla 4. Dosis terapéutica media por fármaco
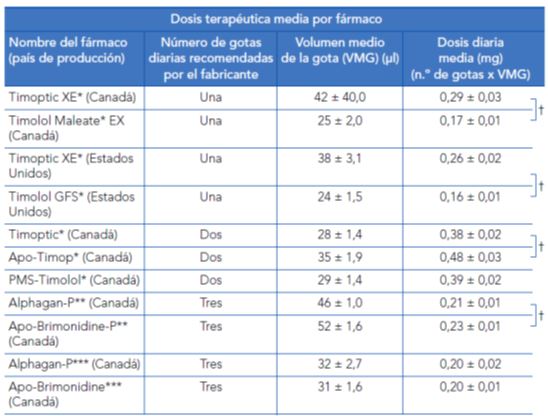
* Timolol maleato solución al 0,5% (6,8mg/ml).
** Brimonidina tartrato al 0,15% (1,5 mg/ml).
*** Brimonidina tartrato al 0,2% (2 mg/ml).
t Los corchetes a la derecha de la tabla indican una diferencia significativa entre las medias (ANOVA) (p < 0,01).
Fuente: Mammo ZN, Flanagan JG, James DF, Trope GE. Generic versus brand-name North American topical glaucoma drops. Can J Ophthalmol. 2012;47(1):55-61
Diferencias clínicas entre genéricos y fármacos de marca oftálmicos antiinflamatorios
Se observaron complicaciones corneales después de la instilación de diclofenaco en el postoperatorio de procedimientos oftálmicos rutinarios. Se pensó en un efecto de clase10. Finalmente se relacionó con un genérico de una determinada marca y fue rápidamente retirado del mercado11. No se informó sobre cuál de los componentes del fármaco fue el responsable de las complicaciones corneales.
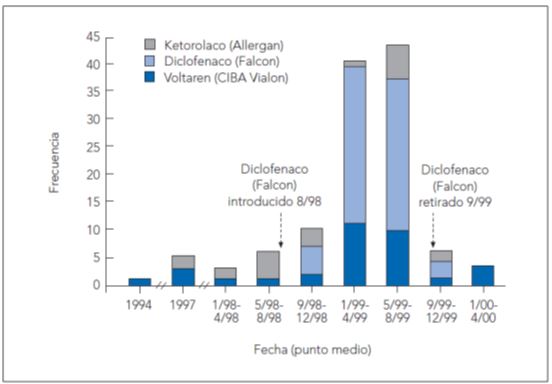
Figura 4. Evolución en el tiempo de todos los casos confirmados de patologia corneal asociados con la administración de antiinflamatorios no esteroideos tópicos.
Fuente: Congdon NG, Schein OD, von Kulatja P, Lubomski LH, Gilbert D, Katz J. Corneal complications associated with tropical ophthalmic use of nonsteroidal antiinflammatory drugs. J Cataract Refract Surg. 2001;27(4):622-31.
El acetato de prednisolona es un fármaco lipofílico, y por ello la preparación original (PredForte®, Allergan) está específicamente elaborada como suspensión. La suspensión de acetato de prednisolona al 1 % requiere que el paciente agite previamente el envase para facilitar la formación de una suspensión homogénea. Los problemas en la suspensión del fármaco genérico producido por Sabex Pharmeceutical (Quebec) exigían al menos 70 sacudidas para que pudiera obtenerse una suspensión homogénea12. El uso de otro genérico producido por Falcon Laboratories (Forth Worth) producía una oclusión del orificio del colirio por lo que parecía ser un precipitado de acetato de prednisolona (12). Tras la instilación del colirio genérico Econopred Plus (Alcon), después de ser agitado cinco veces, se obtenía una concentración del principio activo en cada gota significativamente menor en comparación con el fármaco de marca; un 10 % de su concentración máxima frente al 47 % de PredForte®13.
En resumen, distintos investigadores12,14 han observado en formulaciones genéricas de acetato de prednisolona diferentes problemas relacionados con su carácter de suspensión y la formación de precipitados. Entre ellos, alteración de la homogeneidad de la suspensión, oclusión del orificio del colirio y concentración significativamente menor del principio activo en cada gota.
Genéricos de fármacos oftálmicos antiglaucomatosos
La formulación genérica de la solución de maleato de timolol que se convierte en gel (Falcon Laboratories) fue clasificada por la FDA como AB en ausencia de estudios comparativos entre el fármaco de marca y el genérico, aunque esos productos están formulados con diferentes vehículos de gel de liberación lenta. El tiempo de contacto con la superficie corneal difiere entre ambos fármacos así como los excipientes inactivos y las concentraciones de los conservantes.
Stewart et al. (2002)15 realizaron un estudio comparativo, aleatorio, cruzado, de 12 semanas de duración, entre Timoptic XE® y el correspondiente fármaco genérico en 32 pacientes. La PIO se medía a las 8:00 horas (efecto valle) y a las 2 y 8 horas de la instilación. No se observaron diferencias estadísticamente significativas entre ambos fármacos a las 8:00 horas ni a las 2 horas de la aplicación. Sin embargo, a las 8 horas de la instilación la PIO era de 17,5 ± 3,2 mmHg en el grupo del Timoptic XE® y de 18,9 ± 3,3 en el grupo del fármaco genérico (p = 0,0019). La seguridad así como el tiempo de recuperación de la visión fue similar entre ambos grupos.
Narayanaswamy et al. (2007)16 realizaron un ensayo clínico aleatorizado y cruzado de 24 semanas de duración realizado en India. Se trató secuencialmente a pacientes con hipertensión ocular o glaucoma de ángulo abierto con un genérico de latanoprost comercializado en dicho país (Latoprost® comercializado por Sun Pharmaceuticals) o con latanoprost de referencia (Xalatan® comercializado por Pfizer Ophtalmics). Se incluyeron un total de 30 pacientes. Doce pacientes fueron tratados durante un primer período de 12 semanas con Xalatan® mientras que los 18 pacientes restantes recibieron Latoprost®. En un segundo período, desde la semana 13 a la 24 del estudio, los pacientes tratados durante el primer período con Xalatan® recibieron Latoprost® y los que habían recibido Latoprost® cambiaron a Xalatan®.
Al final del primer período de tratamiento, se observó que en los pacientes tratados con Xalatan®, la PIO había disminuido desde unas cifras basales de 23,64 ± 3,13 mmHg a 14,29 ± 1,61 mmHg (descenso de un 39 %). En los pacientes tratados con Latoprost® la reducción fue desde 22,74 ± 2,47 mmHg a 16,98 ± 2,49 mmHg (descenso de un 25 %). En ambos grupos, todos los pacientes analizados presentaron, tras 12 semanas de tratamiento, cifras de PIO inferiores a 21 mmHg y por tanto habrían alcanzado valores que se consideran dentro del rango de la normalidad. En el segundo período, los pacientes que cambiaron de Xalatan® a Latoprost® presentaron al final de las 24 semanas unas cifras de PIO de 15,36 ± 1,71 mmHg y los que cambiaron de Latoprost® a Xalatan® de 16,09 ± 1,49 mmHg. No hubo diferencias significativas en la incidencia de hiperemia conjuntival ni de ningún otro efecto adverso.
Tabla 5. Presion intraocular (en mmHg) en ambos grupos en la duodécima semana
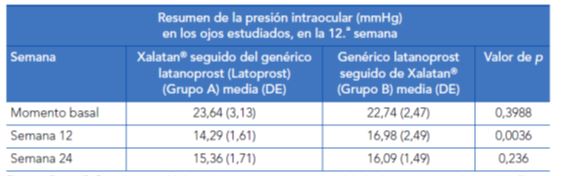
Fuente: Desai C, Rajadhyaksha V. A randomized, crossover, open label pilot study to evaluate the efficacy and safety of Xalatan in comparison with generic Latanoprost (Latoprost) in subjects with primary open angle glaucoma or ocular hypertension. Indian J Ophthalmol. 2007;55(2):127-31.
Los autores del estudio compararon la magnitud del descenso en la PIO al final del primer período y hallaron una diferencia estadísticamente significativa entre los dos grupos estudiados. Al final del segundo período no existían diferencias significativas entre los dos tratamientos.
El fármaco genérico tenía un pH más alto y mayor concentración de partículas. Esas diferencias pueden afectar potencialmente a la estabilidad así como a la liberación del principio activo en el ojo.
Strmen et al. (2010)17 realizan un estudio comparativo y aleatorizado, realizado en la República Checa, publicado en eslovaco y del que sólo disponemos del resumen. Se compara la eficacia en la reducción de la PIO de una nueva formulación de latanoprost (Unilat) frente a Xalatan®.
Se estudió a 77 pacientes, siendo la variable primaria de eficacia el descenso medio de la PIO y la variable secundaria el porcentaje de pacientes con valores de PIO inferiores a 21 mmHg al final del estudio. De acuerdo con lo comunicado por los autores en el resumen, el estudio demostró la no inferioridad del nuevo producto con el de referencia.
¿Cuál va a ser la repercusión en la clínica diaria del cambio a fármacos genéricos de medicamentos antiglaucomatosos?
Noecker y Simmons (2010)18,19 consideran que aunque los fabricantes de formulaciones genéricas cumplan todos los estándares exigidos por la FDA, no se conoce cuál va a ser la respuesta de un paciente individual a un determinado fármaco. Mientras algunos pacientes toleran cambios en sus gotas con mínimas molestias, otros son extremadamente sensibles y pueden referir incluso pequeños cambios relacionados con los conservantes, el pH, la tonicidad u otros componentes. Esto puede llevar a un menor cumplimiento o a manifestar distintos efectos adversos.
Los pacientes generalmente prueban el fármaco que prescribimos. Si causa molestias o genera efectos adversos, suspenden su administración y no lo refieren habitualmente hasta el siguiente control. En ocasiones, los efectos adversos son medibles en términos de signos, pero en otras se manifiestan como quejas sistémicas vagas que surgen con el tiempo.
Los autores consideran que el cambio de un fármaco original por un genérico puede ser algo así como un proceso de ensayo y error19. Por esta razón, cuando un paciente cambia a un genérico, hay que reducir el tiempo entre las visitas de seguimiento para garantizar que el nuevo medicamento está funcionando como se espera y el paciente no tiene molestias.
Ante la pregunta típica y esperable: «Doctor, he recibido una notificación de mi médico, de mi farmacéutico, de mi seguro y me dicen que tengo que cambiar a un genérico. ¿El fármaco genérico es tan bueno como el fármaco de marca?»
Noecker y Simmons19 adoptan posiciones diferentes según el estado clínico del paciente
1) El paciente ha sido diagnosticado recientemente de glaucoma y la enfermedad
está en un estadio inicial. En este caso somos más proclives a sugerir que pruebe o inicie tratamiento con el fármaco genérico.
2) En pacientes con glaucoma avanzado, especialmente si están ahora bien controlados y han probado con anterioridad múltiples fármacos hasta alcanzar el estado actual, somos mucho más reacios al cambio. Los autores refieren que según su experiencia, cuando los pacientes cambian de fármaco, puede ser más difícil volver a alcanzar el control de la enfermedad.
Por su parte Cantor (1997)20 considera que cuando se cambia a un fármaco genérico, ni el paciente ni el médico tienen la seguridad de que el nuevo fármaco será bien tolerado o efectivo. El autor señala que como médicos no podemos saber si el fármaco genérico penetra en el ojo igual o de forma diferente al fármaco de marca y refiere que cuando los pacientes nos preguntan acerca de la sustitución del fármaco de marca por un genérico, todo lo que como médicos podemos decir honestamente es que este nuevo fármaco genérico no ha sido estudiado, analizado o comparado frente al fármaco original y por ello no sabemos si va a actuar de la misma forma, o lo que es lo mismo, si son igual de efectivos. Concluye que los temores que suscitan los fármacos genéricos sólo desaparecerán cuando se promuevan y realicen estudios comparativos con los fármacos originales en relación con la eficacia, seguridad y confort del paciente.
Noecker y Simmons19 refieren que «No hay duda de que estamos cambiando hacia un mundo totalmente genérico y en la medida en que nuestros pacientes se adaptan a medicinas diferentes, nosotros debemos mantener una actitud vigilante para valorar la eficacia y la seguridad. Eso no significa decir a los pacientes que no usen fármacos genéricos, sino que debemos conocer sus efectos para asegurar a nuestros pacientes que están obteniendo la reducción de costes que ellos esperan, sin sacrificar eficacia y seguridad en forma de efectos colaterales o tolerancia.»
Por último, algunos autores consideran que si la principal y única ventaja de los fármacos genéricos es el coste, al realizar un análisis de costes no se debería comparar únicamente el precio del fármaco sino que deberían ser tenidos en cuenta los siguientes factores 1) Tamaño de la gota y capacidad del envase 2) Conveniencia y confort. 3) Repercusión en el cumplimiento. 4) Número potencial de consultas o procedimientos «extras» y 5) Grado de incertidumbre a largo plazo.
*Este artículo es una adaptación del libro realizada por el autor y publicado por
Editorial Glosa, S.L. (Barcelona)
ISBN: 978-84-7429-572-6
¿Son iguales los genéricos que los fármacos de marca?
Pedro Corsino Fernández Vila